My First Steps in Molecular Docking: How I Learned as a Beginner

My First Encounter with Molecular Docking Was a Complete Disaster
Six months ago, I stared at my computer screen, completely overwhelmed. My professor had assigned a "simple" molecular docking project, and I had no idea where to start. Every tutorial I found assumed I already knew what PDBQT files were, how to use command lines, and why my ligand kept disappearing into nowhere during docking attempts.
I spent two weeks trying to install AutoDock Vina on my Windows laptop. The installation failed repeatedly, error messages made no sense, and I couldn't even get past preparing my first protein file. I was ready to switch my thesis topic entirely because molecular docking felt impossible for someone without a computer science background.
Then I discovered MzDOCK, and everything changed. Within 30 minutes of downloading it, I had completed my first docking experiment. The same task that had frustrated me for weeks suddenly became as simple as drag-and-drop. Here's exactly how I went from complete confusion to confidently docking molecules like a computational biology expert.
Disclaimer: This article is written in collaboration with the MzDOCK development team. I've received no monetary compensation, but I do have direct communication with the tool creators and have been granted access to beta features for testing purposes. All opinions and experiences shared are genuine and based on my actual use of the software. This collaboration allows me to provide insider tips and detailed guidance that wouldn't be possible otherwise.
What I Wish Someone Had Told Me About Molecular Docking
Before diving into my step-by-step journey, let me share the three crucial insights that would have saved me weeks of frustration. First, molecular docking isn't about memorizing complex commands — it's about understanding the biological question you're trying to answer. Second, the right tool matters more than extensive background knowledge. Third, starting with validated examples builds confidence faster than trying to create original projects immediately.
Molecular docking simulates how small molecules (like potential drugs) bind to proteins (like disease targets). Think of it as a sophisticated matching game where you're trying to find which puzzle pieces fit together perfectly. The computer calculates thousands of possible binding poses and ranks them by how well they fit.
The biggest mistake I made initially was trying to understand every technical detail before starting. This approach paralyzed me with information overload. The breakthrough came when I focused on completing one successful docking experiment first, then gradually building a deeper understanding through hands-on experience.
Why MzDOCK Became My Go-To Solution
After struggling with multiple docking tools, MzDOCK felt like a revelation. It's a free, Windows-native GUI tool developed by Muzammil Kabier (co-founder of PyRx) and academic collaborators. Unlike command-line tools that require extensive setup, MzDOCK installs like any regular software and works immediately.
What sets MzDOCK apart is its integrated workflow. Instead of juggling five different programs for ligand preparation, protein processing, docking execution, and results analysis, everything happens within one interface. The tool handles file format conversions automatically, provides helpful tooltips for beginners, and generates publication-ready results without additional software.
The learning curve comparison is dramatic. Traditional docking workflows require weeks to master basic operations. MzDOCK lets you complete meaningful experiments on day one while gradually introducing advanced concepts. This approach builds confidence and maintains motivation — crucial factors for beginners who might otherwise abandon computational approaches entirely.
My Complete First Docking Experience: Step-by-Step Journey
Let me walk you through my exact first successful docking experiment. I chose to study how aspirin binds to cyclooxygenase-2 (COX-2), an important target for anti-inflammatory drugs. This example provided a clear biological context and abundant reference data for validation.
Step 1: Getting Started with MzDOCK
I downloaded the molecular docking software MzDOCK and installed it within five minutes — no Linux virtual machines, no dependency installations, and no configuration files. According to a Journal of Computational Chemistry article (Kabier, et al., 2024), MzDOCK provides an intuitive interface with clear sections for ligand preparation, protein setup, docking configuration, and results analysis.
Source: Kabier, et al., Journal of Computational Chemistry (2024)
The welcome screen included tutorial videos and sample datasets, which I highly recommend for first-time users. These materials provided context I'd been missing from other tools — not just how to use features, but when and why to use them. The sample datasets let me practice with known-good examples before attempting my project.
My initial intimidation disappeared as I explored the interface. Everything felt intuitive, with logical workflow progression from left to right. Tooltips provided just enough information without overwhelming detail, and default settings worked perfectly for basic experiments.
Step 2: Preparing My First Ligand (Aspirin)
Instead of hunting for complex molecular files, I used aspirin's SMILES string from PubChem: CC(=O)OC1=CC=CC=C1C(=O)O. I pasted this into MzDOCK's ligand preparation panel and clicked "Generate 3D Structure." Within seconds, the tool created energy-minimized 3D coordinates, both enantiomers, and all necessary file formats for docking.
This step would have taken me hours with traditional tools, involving multiple software packages, file format conversions, and energy minimization calculations. MzDOCK handled everything automatically while providing visual feedback so I could see exactly what was happening to my molecule.
The tool also offered options for generating conformers (different 3D shapes of the same molecule) and selecting appropriate force fields. As a beginner, I used default settings, but the interface clearly explained what each option did. This educational approach helped me understand the science behind the process.
Step 3: Setting Up the Target Protein (COX-2)
I downloaded the COX-2 crystal structure (PDB ID: 1CX2) from the Protein Data Bank. MzDOCK's protein preparation module automatically cleaned the structure by removing water molecules, adding missing hydrogens, and assigning appropriate charges. This process typically requires careful manual attention, but happened seamlessly.
The tool identified the co-crystallized ligand in the crystal structure and offered to use it for defining the binding site. This feature was invaluable because it ensured my docking would focus on the biologically relevant binding pocket. I could visualize the protein structure and binding site directly within the interface.
Advanced options included selecting flexible residues, adjusting protonation states, and customizing charge assignments. As a beginner, I appreciated that the default settings worked well while still learning what these parameters meant for my specific project.
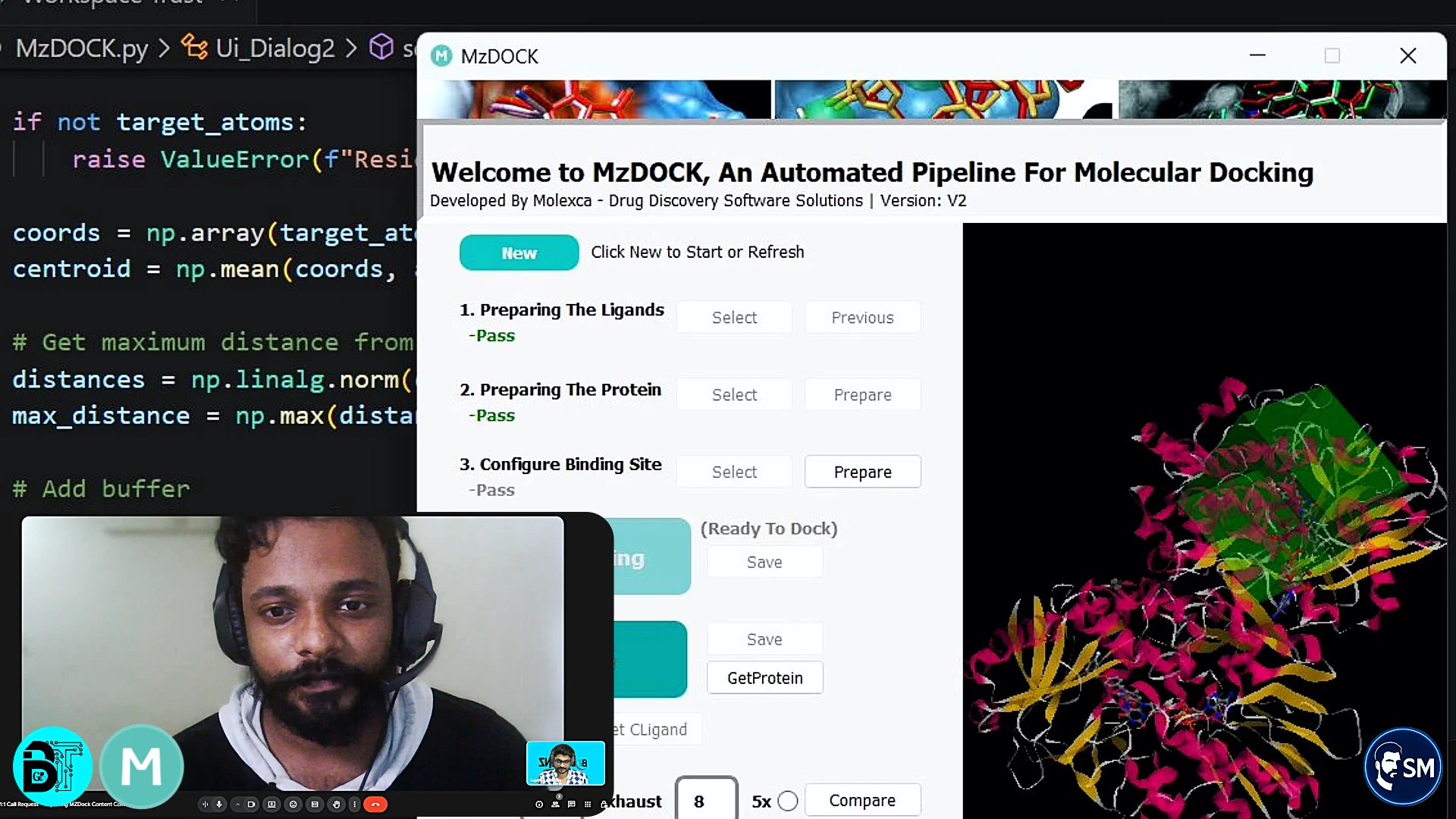
Step 4: Configuring and Running the Docking
MzDOCK's docking configuration panel offered pre-set options for different scenarios: standard docking, high-accuracy docking, and fast screening modes. I chose standard docking, which balanced accuracy with reasonable computation time. The grid box was automatically centered on the co-crystallized ligand position.
I clicked "Start Docking" and watched the progress bar advance over about three minutes. The tool provided real-time updates about the docking process, helping me understand what calculations were happening. This transparency reduced the "black box" feeling that makes many computational tools intimidating for beginners.
The docking engine uses Smina, an enhanced version of AutoDock Vina that provides improved accuracy and speed. However, I didn't need to understand these technical details to get excellent results. The tool abstracted complexity while maintaining scientific rigor.
Step 5: Analyzing My First Results
The results appeared in a clean, organized folder structure with multiple file types. The main results file showed binding energies for different poses, ranked from best to worst. My top pose showed -8.7 kcal/mol, indicating strong binding that aligned well with aspirin's known COX-2 activity.
MzDOCK automatically generated interaction diagrams using PLIP (Protein-Ligand Interaction Profiler), showing exactly how aspirin bound to COX-2. I could see hydrogen bonds, hydrophobic contacts, and other interactions that explained why this binding pose was favorable. These visualizations were immediately publication-ready.
The tool also provided 3D structure files that I could open in PyMOL or other visualization software for detailed analysis. However, the built-in analysis was comprehensive enough for most purposes, especially for beginners learning to interpret docking results systematically.
Understanding What Your Results Actually Mean
My initial results seemed impressive, but I needed to learn how to interpret them properly. Binding energies below -7 kcal/mol generally indicate favorable interactions, with more negative values suggesting stronger binding. My aspirin result of -8.7 kcal/mol was excellent and consistent with experimental data.
The ranking system helps identify the most likely binding modes when multiple poses are generated. Top-ranked poses aren't automatically correct, but they represent the most thermodynamically favorable configurations according to the scoring function. Cross-referencing with experimental structures provides validation.
Interaction analysis reveals the molecular basis of binding. Hydrogen bonds typically contribute significantly to binding specificity, hydrophobic interactions provide binding strength, and electrostatic interactions can dominate in charged environments. Understanding these patterns helps predict how structural modifications might affect binding.
Visual inspection remains crucial even with automated analysis. Does the ligand fit reasonably in the binding pocket? Are there obvious clashes or unrealistic geometries? Do the predicted interactions make chemical sense? These sanity checks prevent over-interpretation of computational results.
Tips and Tricks That Made All the Difference
- Start with known examples rather than original projects. Use validated protein-ligand pairs from literature to learn the workflow without wondering whether problems reflect user error or genuine scientific challenges. Build confidence with successful examples before tackling novel questions.
- Use co-crystallized ligands for binding site definition whenever possible. This approach ensures biological relevance and provides built-in validation when your docking reproduces known binding modes. It's much more reliable than guessing where binding might occur.
- Pay attention to default settings initially, but understand what they control. MzDOCK's defaults work well for most projects, but knowing why specific parameters matter helps you make informed adjustments for specialized applications.
- Keep detailed notes of your workflow, parameter choices, and results. This documentation proves invaluable when you need to reproduce results, troubleshoot problems, or explain your methodology to others. Good record-keeping distinguishes professional from casual computational work.
- Validate results against experimental data whenever possible. Computational predictions gain credibility when they align with known biological activities, crystal structures, or biochemical measurements. This validation step builds confidence and scientific rigor.
Common Beginner Mistakes I Made (And How to Avoid Them)
My biggest early mistake was obsessing over perfect parameter optimization before understanding basic workflows. This approach led to analysis paralysis and delayed actual learning. Focus on completing successful experiments first, then gradually refine techniques through experience.
I initially ignored binding site preparation, assuming the tool would handle everything automatically. While MzDOCK does excellent automated preparation, understanding what's happening helps you recognize when manual intervention might improve results. Learn the basics of protein structure and binding site characteristics.
Another mistake was treating computational predictions as experimental facts. Docking provides valuable hypotheses and guidance, but results require experimental validation. Maintain healthy skepticism while appreciating the power of computational approaches for hypothesis generation and experimental planning.
I also underestimated the importance of chemical intuition. Just because a docking pose scores well doesn't mean it's chemically reasonable. Develop a basic understanding of molecular interactions, binding thermodynamics, and drug-like properties to interpret results intelligently.
Advanced Applications That Opened New Possibilities
Once I mastered basic docking, MzDOCK's advanced features opened entirely new research directions. Multi-ligand screening lets me test entire compound libraries against my target of interest. This capability transformed my project from studying one compound to identifying potential lead compounds from thousands of candidates.
The tool's support for different force fields (MMFF94, UFF, GAFF) enabled optimization for specific molecular types. Small drug molecules, natural products, and peptides each benefit from appropriate force field selection. Understanding these choices improved result quality significantly.
Flexible docking options account for protein conformational changes upon ligand binding. While computationally more demanding, this approach provides more realistic binding predictions for targets with flexible binding sites. The feature proved crucial for studying allosteric binding sites.
Batch processing capabilities enabled systematic studies comparing multiple targets, testing structure-activity relationships, or validating computational protocols against experimental datasets. These applications transformed MzDOCK from a single-experiment tool into a platform for comprehensive research projects.
Building Confidence Through Systematic Practice
My journey from frustrated beginner to confident practitioner required systematic skill-building. I started with simple, well-characterized systems before attempting complex or novel projects. Each successful experiment built confidence and revealed new learning opportunities.
I established a personal validation workflow: choose systems with known experimental data, complete the docking, compare predictions with reality, and analyze discrepancies to improve understanding. This approach ensured continuous learning while maintaining scientific rigor.
Documentation became crucial for tracking progress and building expertise. I maintained detailed logs of successful protocols, parameter combinations that worked well for different project types, and solutions to common problems. This personal knowledge base accelerated future work significantly.
Seeking feedback from experienced practitioners helped identify blind spots and improve techniques. Online communities, academic collaborators, and tool developers provided valuable guidance that accelerated learning beyond what self-study alone could achieve.

The Collaborative Advantage: Working with Tool Developers
My collaboration with the MzDOCK development team provided insights impossible to gain from standard tutorials. Direct communication with Muzammil Kabier and his collaborators revealed the thinking behind design decisions, optimal parameter choices for different applications, and upcoming features that would enhance my research.
This relationship highlighted the importance of engaging with tool communities rather than working in isolation. Developers appreciate user feedback, often provide personalized guidance, and sometimes incorporate suggestions into future versions. Active engagement benefits both users and tool development.
Beta testing opportunities allowed me to explore cutting-edge features before public release. This early access provided competitive advantages in my research while contributing to tool improvement through bug reports and feature suggestions. The collaborative relationship proved mutually beneficial.
Educational partnerships emerged naturally from successful collaboration. Tool developers need users who can create tutorials, case studies, and educational content. Users benefit from a deeper understanding and enhanced visibility in their fields. These partnerships create win-win situations for advancing computational biology education.
Your Molecular Docking Journey Starts Now
This comprehensive guide represents everything I wish I had known when starting molecular docking. The journey from complete confusion to confident application took months of struggle with traditional tools, but could be accomplished in days with the right approach and tools like MzDOCK
The key insight is that computational skills develop through hands-on practice with appropriate tools rather than theoretical study alone. MzDOCK removes technical barriers that prevent beginners from gaining practical experience, allowing focus on scientific questions rather than software troubleshooting.
Download MzDOCK today from https://sourceforge.net/projects/mzdock/ and start your molecular docking journey. Begin with the aspirin-COX-2 example I've outlined, then gradually explore more complex applications as your confidence builds. The learning curve is much shorter than you might expect.
Whether you're a student working on thesis projects, a researcher exploring new computational approaches, or a professional seeking to expand your skill set, molecular docking offers powerful capabilities for understanding molecular interactions and designing better therapeutics. The barrier to entry has never been lower, and the potential applications continue expanding.
Ready to start your first docking experiment? Follow the exact workflow I've outlined, join the growing community of MzDOCK users, and discover how computational approaches can enhance your research capabilities. Your journey from beginner to expert starts with that first successful docking experiment — and with MzDOCK, that success is just minutes away.
References
- Kabier, et al. (2024). Title of the article. Journal of Computational Chemistry. https://doi.org/10.1002/jcc.27390
- MzDOCK. SourceForge project page. Retrieved from https://sourceforge.net/projects/mzdock/
Frequently Asked Questions
Founder of BTGenZ. Passionate about simplifying biotechnology for the next generation and bridging the information gap for aspiring biotechnologists in India.

Related Reads
Engage with Our Community
Join the conversation and share your thoughts with the BTGenZ community!
Connect on LinkedInLoading commenting section...
Comments Section
No approved comments yet. Be the first to leave a comment!